Adapter trimming
In the next step, Trim Galore finds and removes adapter sequences from the 3' end of reads.
Adapter auto-detection
Section titled “Adapter auto-detection”If no sequence was supplied, Trim Galore will attempt to auto-detect the adapter that has been used. For this it will analyse the first 1 million sequences of the first specified file and count exact substring matches against a set of standard adapter probes:
Illumina: AGATCGGAAGAGC (13 bp)Small RNA: TGGAATTCTCGG (12 bp)Nextera: CTGTCTCTTATA (12 bp)BGI/DNBSEQ: AAGTCGGAGGCCAAGCGGTCTTAGGAAGACAA (32 bp; v2.x addition)If no adapter contamination can be detected within the first 1 million sequences, or in case of a tie, Trim Galore defaults to --illumina. The auto-detection results are shown on screen and printed to the trimming report for future reference.
In v2, auto-detection runs per pair rather than once per invocation, so a glob like trim_galore --paired *fastq.gz correctly handles multiple samples with different library types or 2-colour/4-colour chemistries.
The Stranded Illumina adapter (ACTGTCTCTTATA) is intentionally not auto-detected: it differs from the Nextera adapter by a single A-tail base, so probing both would produce constant ambiguous ties. Pass --stranded_illumina explicitly when working with those libraries.
Multiple adapter sequences
Section titled “Multiple adapter sequences”Multiple adapters can be specified in three equivalent ways. The cleanest (v2.x) is simply to repeat -a and/or -a2:
trim_galore -a AGCTCCCG -a TTTCATTATAT -a TTTATTCGGATTTAT -n 3 input.fastq.gztrim_galore --paired -a2 AGCTAGCG -a2 TCTCTTATAT -a2 TTTCGGATTTAT -n 3 R1.fq.gz R2.fq.gzThe v0.6.x embedded-string form is still accepted for backwards compatibility:
trim_galore -a " AGCTCCCG -a TTTCATTATAT -a TTTATTCGGATTTAT" input.fastq.gztrim_galore -a2 " AGCTAGCG -a TCTCTTATAT -a TTTCGGATTTAT" --paired R1.fq.gz R2.fq.gzOr load adapters from a FASTA file:
trim_galore -a "file:./adapters.fa" input.fastq.gztrim_galore -a "file:./r1_adapters.fa" -a2 "file:./r2_adapters.fa" --paired R1.fq.gz R2.fq.gzFor all three forms, adding -n 3 lets Trim Galore strip up to three adapter occurrences from each read. This is useful when adapter contamination can appear multiple times in the same read. Standard trimming does not require -n. More information can be found in Issue 86.
Single-base expansion (A{10} to AAAAAAAAAA) is also supported for both -a and -a2, matching Perl v0.6.x syntax.
Manual adapter sequence specification
Section titled “Manual adapter sequence specification”You can override auto-detection by passing a sequence directly via -a, or by using one of the named presets: --illumina, --nextera, --small_rna, --stranded_illumina, or --bgiseq (run --help for one-line descriptions). The first 13 bp of the standard Illumina adapter (AGATCGGAAGAGC) cover most TruSeq, Sanger iTag, and similar kits, and sit on both sides of paired-end inserts before the index sequence — for normal sequencing, --illumina or auto-detection is enough.
Stringency: why the 1 bp default is correct
Section titled “Stringency: why the 1 bp default is correct”--stringency sets the minimum adapter overlap required to trim. The default is 1 bp, which looks extreme but is deliberate: in bisulfite-seq, even a few residual adapter bases can cause mis-alignments and incorrect methylation calls, or push the read past the aligner's mismatch budget so the entire read drops out. The cost is occasionally trimming a true genomic base, but in directional bisulfite libraries only the 4th–5th adapter bases overlap with the methylation-callable region, so the trade tilts firmly toward stringent trimming.
| Before adapter trimming | After adapter trimming |
|---|---|
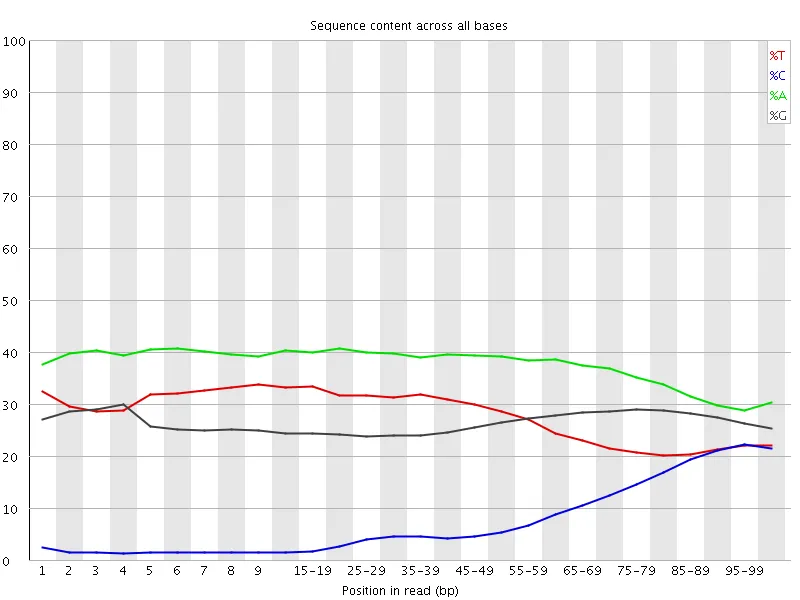 | 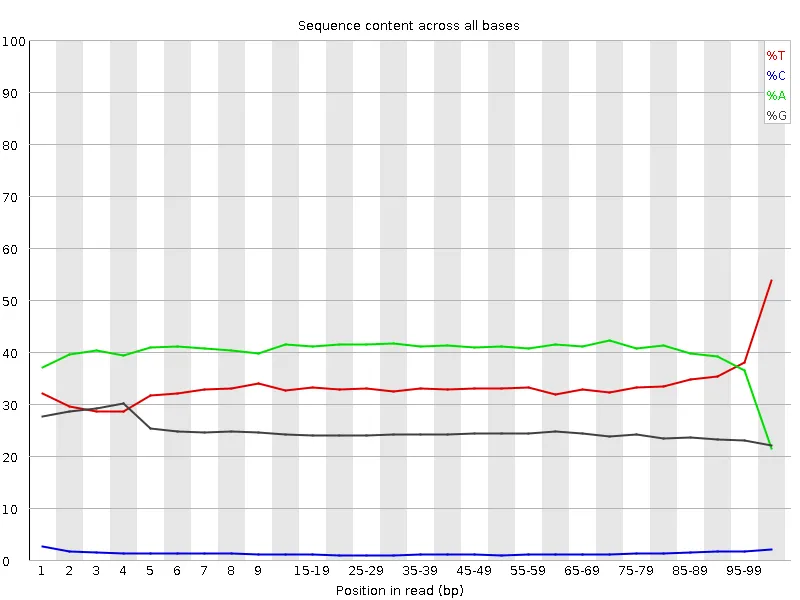 |
C content rises from ~1% at the start of reads to ~22% by the 3' end without trimming. Trim Galore removes the contamination cleanly. The sharp drop in A at the very last position is the visible footprint of --stringency 1 doing its job — single trailing A's are removed by design.
Trimming error rate
Section titled “Trimming error rate”The default error tolerance for adapter alignment is -e 0.1 (10%). Lowering this is rarely useful; raising it can recover more contamination at the cost of more false positives.
Already-trimmed data
Section titled “Already-trimmed data”--consider_already_trimmed <INT> suppresses adapter trimming entirely when no auto-detect probe exceeds that count (quality trimming still runs). Useful for feeding already-trimmed data through Trim Galore for QC reporting without over-trimming.
Related flags
Section titled “Related flags”| Flag | Purpose |
|---|---|
-a SEQ / -a2 SEQ | Adapter for read 1 / read 2 (repeatable in v2). |
--illumina / --nextera / --small_rna / --stranded_illumina | Force a specific known adapter. |
--bgiseq | Use the BGI/DNBSEQ adapter (also probed by auto-detect). |
--stringency INT | Minimum adapter overlap (default 1 bp). |
-e FLOAT | Maximum error rate for adapter alignment (default 0.1). |
-n INT | Trim up to N adapter occurrences per read (multi-adapter mode). |
--consider_already_trimmed INT | Skip adapter trimming if no adapter exceeds the count threshold. |