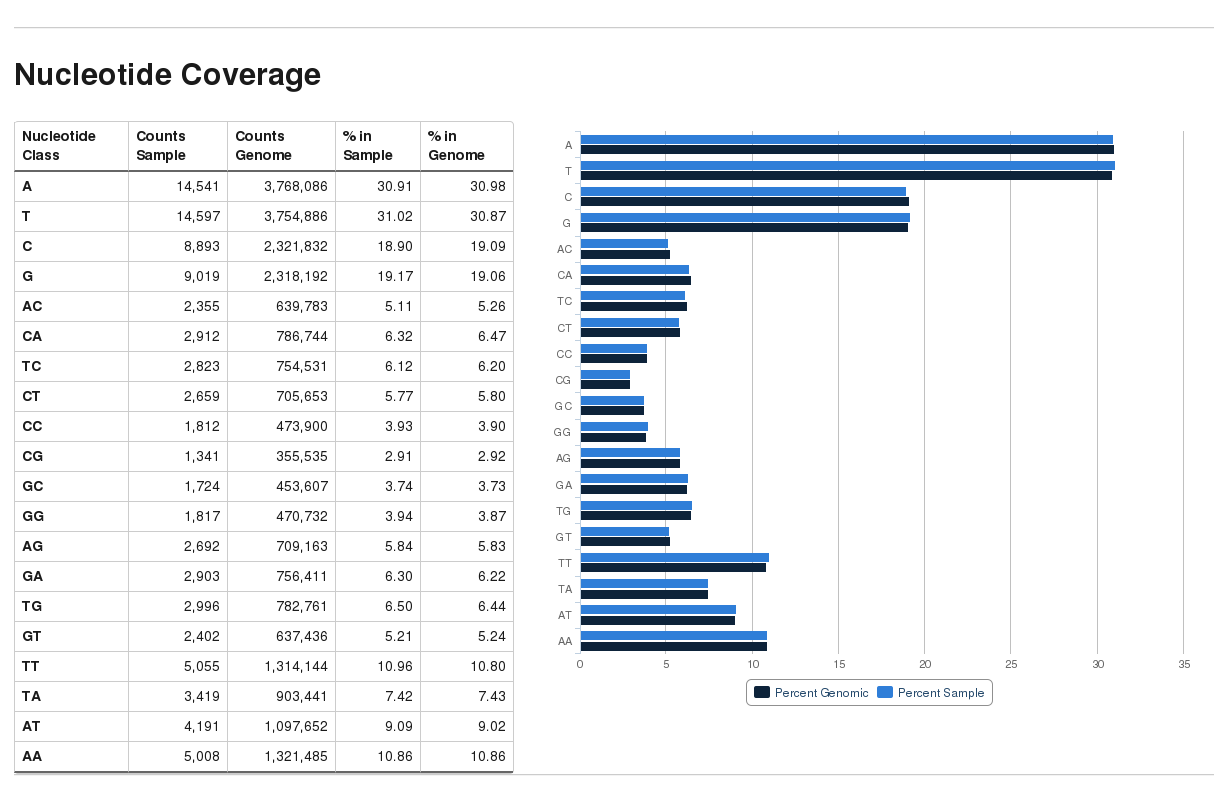
Nucleotide coverage report
The script bam2nuc reads BAM files and calculates the mono- and di-nucleotide coverage of the
reads (using the genomic sequence rather than the observed sequence in the reads themselves)
and compares it to the average genomic sequence composition. Reads harbouring InDels are not
taken into consideration. Mono- or dinucleotides containing Ns are ignored as well.
Usage
Arguments
Aligned BAM files. bam2nuc handles both Bismark single-end and paired-end files (determined automatically).
Note
Both BAM and CRAM files should work as input, but please note that Samtools version 1.2 or higher is required for CRAM files.
Options
--dir
Output directory. Output is written to the current directory if not specified explicitly.
--genome_folder <path>
Enter the genome folder you wish to use to extract sequences from (full path only). Accepted formats are FastA files ending with .fa or .fasta. Specifying a genome folder path is mandatory.
--samtools_path
The path to your Samtools installation, e.g. /home/user/samtools/. Does not need to be specified explicitly if Samtools is in the PATH already
--genomic_composition_only
Only calculate and extract the genomic sequence composition and exit thereafter.
This option will attempt to write the genomic composition table genomic_nucleotide_frequencies.txt to the genome folder or to the output directory instead if that doesn't succeed.
--help
Displays this help message and exits
Genomic composition
Since the calculation of the average genomic (di-)nucleotide composition may take a while, bam2nuc attempts to write out a file called 'genomic_nucleotide_frequencies.txt' to the genome folder if it wasn't there already. The next time bam2nuc is run it will then use this file instead of calculating the average genome composition again. If writing to the genome folder fails (e.g. because of permission issues) it will be written out to the output directory instead.
Output format
bam2nuc writes out a file ending in .nucleotide_stats.txt in the following format (tab-delimited):
(di-)nucleotide count sample percent sample count genomic percent genomic coverage
A 14541 30.91 3768086 30.98 0.004
C 8893 18.90 2321832 19.09 0.004
G 9019 19.17 2318192 19.06 0.004
T 14597 31.02 3754886 30.87 0.004
AA 5008 10.86 1321485 10.86 0.004
AC 2355 5.11 639783 5.26 0.004
AG 2692 5.84 709163 5.83 0.004
AT 4191 9.09 1097652 9.02 0.004
CA 2912 6.32 786744 6.47 0.004
CC 1812 3.93 473900 3.90 0.004
CG 1341 2.91 355535 2.92 0.004
CT 2659 5.77 705653 5.80 0.004
GA 2903 6.30 756411 6.22 0.004
GC 1724 3.74 453607 3.73 0.004
GG 1817 3.94 470732 3.87 0.004
GT 2402 5.21 637436 5.24 0.004
TA 3419 7.42 903441 7.43 0.004
TC 2823 6.12 754531 6.20 0.004
TG 2996 6.50 782761 6.44 0.004
TT 5055 10.96 1314144 10.80 0.004
This file is picked up and plotted by bismark2report automatically if found in the folder in the following manner: